
3. LA DIAGNOSI: I TEST GENETICI
Pamela Magini, Giovanna Cenacchi, Marco Seri
Le malattie genetiche mendeliane sono patologie rare, causate da un difetto del patrimonio genetico ereditario, e sono classificate come malattie cromosomiche, se dovute ad alterazioni del numero o della struttura dei cromosomi, o malattie geniche, se causate da mutazioni del DNA che alterano la sequenza dei geni e, di conseguenza, la struttura o la funzione delle proteine codificate. Malattie più comuni, che hanno alla base l’interazione tra fattori genetici di suscettibilità e fattori di rischio ambientali, sono definite multifattoriali o complesse.
Grazie al completamento del Progetto Genoma Umano all’inizio degli anni Duemila e allo sviluppo di tecnologie sempre più avanzate, le conoscenze sulle malattie genetiche sono progredite molto rapidamente, permettendo alla ricerca nell’ambito della genetica medica di determinare il ruolo di specifici geni, in oltre 6.000 malattie diverse (Lander et al. 20011Lander, E. S., et al. – International Human Genome Sequencing Consortium (2001) Initial sequencing and analysis of the human genome, Nature, 409(6822): 860- 921.; Venter et al. 20012Venter, J. C., et al. (2001) The sequence of the human genome, Science, 291(5507): 1304-1351.; International Human Genome Sequencing Consortium 20043International Human Genome Sequencing Consortium (2004) Finishing the euchromatic sequence of the human genome, Nature, 431: 931-945.; portale OMIM, cfr. Sitografia – Alcuni link di riferimento).
Le informazioni che derivano dall’identificazione del gene alterato, in una specifica malattia genetica, hanno due importanti applicazioni cliniche: la terapia e la diagnosi. Queste informazioni possono favorire la comprensione del meccanismo patogenetico, che determina l’insorgenza della patologia e che può diventare il bersaglio di terapie mirate. Ad oggi, però, le malattie genetiche “curabili” sono un’esigua percentuale, e l’applicazione medica più rilevante delle conoscenze ottenute dalla ricerca genetica è la diagnosi. Infatti, i test genetici sono fondamentali per confermare, a livello molecolare, un’ipotesi diagnostica basata su informazioni cliniche, con tutto ciò che ne può derivare in termini di rischi di ricorrenza, prognosi della malattia, sorveglianza clinica ed eventuali terapie personalizzate (ACMG Board of Directors 2015).
Poiché è importante arrivare a una definizione del difetto molecolare nei pazienti affetti da malattie rare, l’utilizzo dei test genetici nella pratica medica è andato aumentando in modo esponenziale, soprattutto negli ultimi anni. La definizione comunemente accettata di test genetico è «l’analisi a scopo clinico di DNA, RNA, cromosomi, proteine, metaboliti o altri prodotti genici per evidenziare genotipi, mutazioni, fenotipi o cariotipi correlati o meno con patologie ereditabili umane» (Holtzman, Watson 1999).
I test genetici presentano alcune peculiarità rispetto ad altri esami di laboratorio, prima fra tutte il fatto che i risultati possono avere ricadute non solo sulla persona che si è sottoposta al test, ma anche sui familiari. È per questo che, normalmente, un test genetico va inserito all’interno di un percorso diagnostico specifico e deve essere accompagnato da una consulenza genetica, che dia ai pazienti indicazioni specifiche circa la sua sensibilità e le sue finalità (Fig. 6).
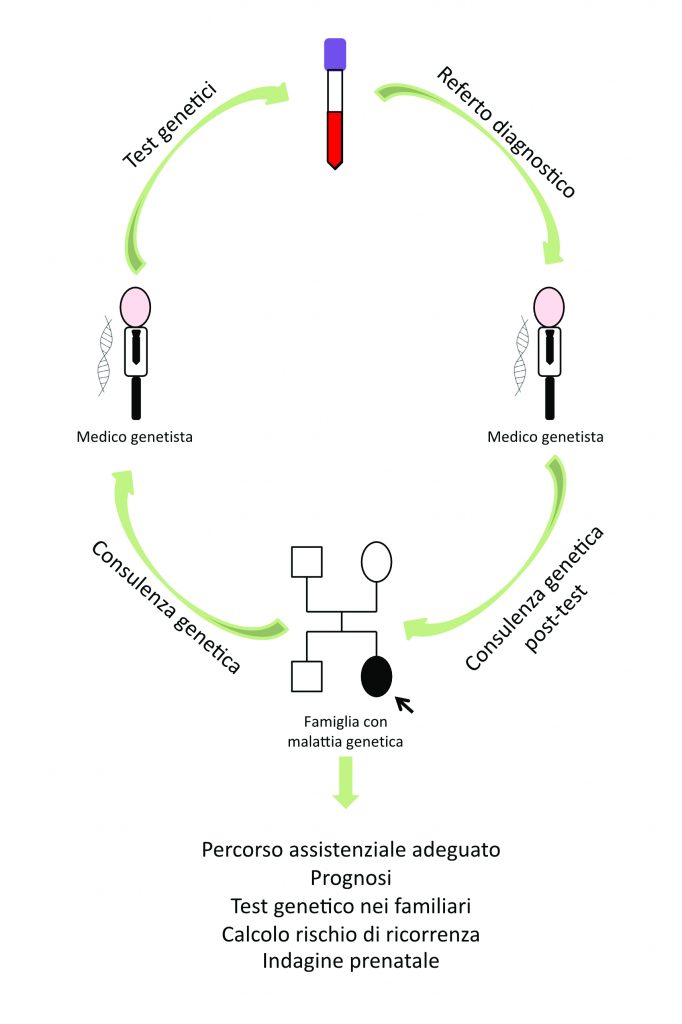
Fig. 6. Quando un medico specialista (pediatra, neuropsichiatra infantile, neurologo, ecc.) sospetta una malattia genetica, generalmente indirizza il paziente da un genetista clinico, il quale lo visita (esame obiettivo), raccoglie i dati anamnestici e la storia familiare. Sulla base dei dati raccolti durante la visita, il genetista clinico può decidere di avviare un test genetico a conferma di un sospetto diagnostico, spiegando al paziente o ai suoi tutori le finalità, le potenzialità e le limitazioni del test stesso, e raccogliendo il consenso informato per la sua esecuzione. Terminata l’indagine genetica, il laboratorio invia il referto al genetista che comunica l’esito al paziente/tutori, durante una consulenza genetica post-test, insieme a tutte le implicazioni che ne derivano per la gestione clinica del paziente e della famiglia.
Proprio in base alle loro finalità e alla tipologia di malattia investigata, i test genetici vengono classificati in diagnostici, presintomatici, predittivi, prenatali (McPherson 20064McPherson, E. (2006) Genetic diagnosis and testing in clinical practice, Clinical medicine & research, 4(2): 123-129). Alcuni test sono indirizzati alla valutazione della suscettibilità genetica per malattie complesse, ma questi spesso hanno una ricaduta limitata in campo medico.
3.1. Test diagnostici
Sono i test che consentono di stabilire una diagnosi, o di confermare un quadro patologico sospettato in un paziente in base alla valutazione clinica. A seguito della diagnosi genetica, è possibile determinare la modalità di trasmissione della malattia e offrire così una consulenza genetica più appropriata, con i rischi di ricorrenza specifici. Possono essere effettuati durante tutto l’arco di vita, ma anche in epoca prenatale. Talvolta l’esito di un test diagnostico consente anche di fare valutazioni circa la prognosi della patologia in esame, se la correlazione tra genotipo e fenotipo è stata caratterizzata in modo approfondito, per cui a determinati genotipi sono stati attribuiti quadri clinici con gradi di gravità e decorso ben definiti. Ad esempio, nella maggior parte delle malattie neurologiche da espansione di triplette, cioè dovute all’aumento del numero di specifiche ripetizioni tri-nucleotidiche, l’età di insorgenza dei sintomi e la gravità della sintomatologia è correlata con il numero di ripetizioni (Paulson 2018). Inoltre, in alcuni casi, la diagnosi molecolare può fornire informazioni utili anche per quanto riguarda la scelta del trattamento terapeutico più efficace. Ad esempio, l’identificazione precoce di mutazioni nel gene SLC2A1 e la tempestiva somministrazione di una dieta chetogenica migliorano notevolmente l’outcome neurologico nei bambini affetti da encefalopatia da deficit di Glut1 (Wang et al. 20025Wang, D., Pascual, J. M., De Vivo, D. (2002) Glucose Transporter Type 1 Deficiency Syndrome, July 30 [updated 2018 Mar 1]. In: M. P. Adam, H. H. Ardinger, R. A. Pagon, et al., eds., GeneReviews®. Seattle (WA): University of Washington.).
3.2. Test presintomatici
Le malattie genetiche, soprattutto con modalità di trasmissione autosomica dominante (dovute a mutazioni di uno dei due alleli di geni localizzati sui cromosomi 1-22, definiti autosomi), che non si manifestano alla nascita ma più tardivamente, anche in età avanzata, vengono definite a insorgenza tardiva. Questo tipo di patologie apre una problematica rilevante nel percorso diagnostico. Infatti, attraverso il test presintomatico, la mutazione causativa identificata in un individuo affetto, che rappresenta il caso indice, può essere ricercata nei familiari asintomatici che, se risultano essere portatori, svilupperanno inevitabilmente la malattia nel corso della loro vita.
Il test presintomatico viene eseguito senza limitazioni per i minorenni, quando l’esito può consentire di ridurre morbilità e/o mortalità, grazie alla disponibilità di forme di prevenzione secondaria o adeguate terapie. Al contrario, il test genetico presintomatico viene limitato ai maggiorenni, avviati a un percorso articolato e gestito da un team multidisciplinare che prevede la presenza del genetista, dello psicologo e dello specialista del caso, quando il risultato non si tradurrebbe in una migliore capacità di gestione clinica, come accade nella maggior parte dei casi.
Tuttavia, non ci sono preclusioni a priori per l’esecuzione del test. Il percorso di supporto servirà al paziente per capire se sottoporsi al test genetico possa essere di aiuto, qualora i risultati forniscano informazioni utili nell’effettuare scelte su alcuni importanti aspetti della vita (maternità/paternità, lavoro, ecc.) oppure, al contrario, se un eventuale risultato positivo del test, che preveda l’insorgenza della malattia, possa essere un peso troppo grande da sopportare, incidendo negativamente sulla sua vita, nel periodo in cui non presenta ancora i sintomi della malattia. Molte patologie, per le quali sono disponibili test presintomatici, sono di ambito neurologico, tra cui l’esempio più classico è quello della corea di Huntington (Quaid 20176Quaid, K. A. (2017) Genetic testing for Huntington disease, Handbook of Clinical Neurology, 144: 113-126. Doi: 10.1016/B978-0-12-801893-4.00010-9.).
3.3. Test predittivi
L’insorgenza di una percentuale relativamente piccola di tumori definiti familiari, come il carcinoma familiare della mammella e dell’ovaio o la sindrome di Lynch (predisposizione a sviluppare il tumore del colon non-poliposico), è dovuta alla mutazione di singoli geni (Garber, Offit 2005). Tuttavia, nella grande maggioranza dei casi, un’alterazione di tipo ereditario in un gene associato al cancro rappresenta solo uno dei potenziali fattori implicati nello sviluppo della malattia ed è, quindi, associata a una maggiore predisposizione alla malattia. Un punto critico è, quindi, la valutazione del valore predittivo del test genetico.
I test predittivi risultano particolarmente importanti, in quanto l’eventuale identificazione di soggetti sani ad alto rischio genetico di cancro comporta la necessità di decidere se intraprendere delle misure di prevenzione. L’approccio alla prevenzione è tuttavia molto complesso, in quanto la disponibilità di misure efficaci varia notevolmente a seconda del tipo di patologia.
3.4. Test prenatali
Durante la diagnosi prenatale, i test genetici possono essere impiegati per identificare alcune patologie genetiche a carico del prodotto del concepimento. Alcuni test sono effettuati senza indicazione specifica (in particolare l’analisi cromosomica, soprattutto nelle donne che hanno superato una certa età, in cui il rischio di generare figli con anomalie cromosomiche è relativamente elevato), altri sono rivolti a specifiche patologie genetiche presenti in famiglia (prevalentemente nel caso di genitori portatori eterozigoti di malattie genetiche recessive, o nel caso di una madre portatrice di un difetto legato al cromosoma X).
Alcuni esempi sono: l’analisi citogenetica per individuare anomalie cromosomiche (ad esempio, l’identificazione della trisomia 21 nella sindrome di Down), la ricerca di mutazioni del gene CFTR in feti a rischio di fibrosi cistica, l’identificazione di espansioni del gene FMR1 in feti concepiti da una donna portatrice di una premutazione.
3.5. Nuove tecnologie applicate alla genetica medica
Nel corso degli anni, il continuo sviluppo di nuove tecnologie per l’analisi del DNA ha consentito di individuare un numero crescente di geni, con un ruolo nella patogenesi di malattie genetiche, e conseguentemente ha portato a un notevole miglioramento della diagnosi nell’ambito della genetica medica, grazie all’aumento della sensibilità e della rapidità nell’identificazione sia di anomalie numeriche/strutturali dei cromosomi, sia di alterazioni della sequenza dei geni (Boycott et al. 2013; Durmaz et al. 2015) come mostrato in Fig. 7.
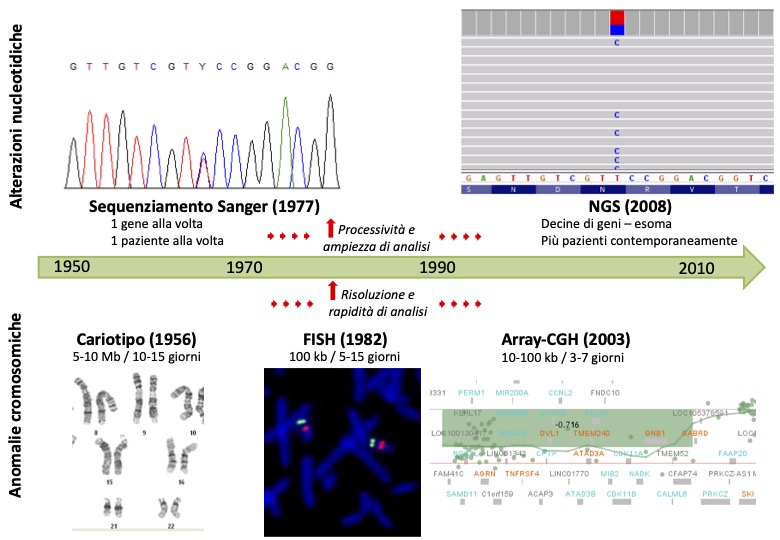
L’analisi del cariotipo (citogenetica classica), che attraverso l’osservazione al microscopio dell’intero assetto diploide dei cromosomi (due copie di ciascun cromosoma, una paterna e una materna) di un individuo con- sente di rilevare anomalie cromosomiche di grandi dimensioni (> 5-10 Mb), è stato il principale test diagnostico dagli anni Cinquanta agli anni Ottanta, quando a esso sono state affiancate tecniche molecolari, quali la FISH (Fluorescence In Situ Hybridization), in cui l’utilizzo di una sonda di DNA fluorescente, complementare alla regione genomica di interesse, ha permesso di osservare possibili delezioni o duplicazioni (Copy Number Variants, CNVs) nei cromosomi del paziente, con una risoluzione migliore rispetto al cariotipo (circa 100 kb).
La vera svolta è avvenuta agli inizi del terzo millennio grazie alla tecnologia array-CGH (array-based Comparative Genomic Hybridization), in cui il DNA del paziente e un DNA di riferimento, marcati con fluorocromi diversi, si ibridano in maniera competitiva a oligonucleotidi (brevi sequenze di nucleotidi) complementari all’intero genoma (DNA contenuto in un organismo, in questo caso l’uomo), adesi su un vetrino che viene scansionato tramite laser, per rilevare i segnali di fluorescenza, calcolare i loro rapporti (DNA paziente/DNA riferimento per ogni oligonucleotide) e determinare il numero di copie corrispondente, con un’elevata risoluzione per l’identificazione di CNVs lungo tutto il genoma (10-100 kb) e con pochi giorni (3-4) di analisi (Pinkel et al. 1998; Albertson, Pinkel 2003). Ad oggi, l’array-CGH viene richiesto come analisi di primo livello per la diagnosi genetica in pazienti affetti principalmente da disabilità intellettiva, isolata o sindromica, ed è frequentemente applicato anche in epoca prenatale con specifiche indicazioni, ad esempio nel riscontro di anomalie ecografiche, o per la definizione di anomalie rilevate all’analisi del cariotipo (Park et al. 20117Park, S.-J., Jung, E. H., Ryu, R.-S., et al. (2011) Clinical implementation of whole-genome array CGH as a first-tier test in 5080 pre and postnatal cases, Molecular Cytogenetics, 4, articolo n. 12.).
Le tecnologie di sequenziamento per l’analisi della sequenza nucleotidica dei geni sono state oggetto di uno sviluppo ancora più rapido (Heather, Chain 20168Heather, J. M., Chain, B. (2016) The sequence of sequencers: The history sequencing DNA, Genomics, 107(1): 1-8.). A partire dalla seconda metà degli anni Settanta fino a pochi anni fa, il metodo Sanger era la tecnica di sequenziamento gold standard e quindi più diffusa nei laboratori. Basandosi sulla separazione tramite elettroforesi capillare di frammenti nucleotidici, amplificati dal DNA del paziente e modificati attraverso l’incorporazione di di-deossiribonucleotidi, definiti terminatori di catena, il metodo Sanger consente l’analisi della sequenza nucleotidica di singoli geni-malattia e l’identificazione di anomalie (sostituzioni, delezioni, inserzioni nucleotidiche) con una processività limitata, con la possibilità di analizzare un solo gene alla volta. Per questo motivo il percorso diagnostico era pressoché proibitivo, soprattutto per le patologie con alta eterogeneità genetica, in cui sono implicati diversi geni. Spesso accadeva che dopo mesi di attesa erano stati sequenziati pochi geni e la causa genetica non era stata identificata.
Lo sviluppo e l’applicazione clinica delle tecnologie di sequenziamento di nuova generazione NGS (Next Generation Sequencing), nel primo decennio degli anni Duemila, hanno migliorato enormemente la diagnosi delle malattie genetiche, sia in termini di efficienza che di efficacia. Grazie a esse oggi è possibile sequenziare più geni contemporaneamente, con un notevole abbattimento dei tempi e dei costi del percorso diagnostico, e offrire al paziente la concreta possibilità di identificare il difetto genetico alla base della sua patologia (Jamuar, Tan 20159Jamuar, S. S., Tan, E.-C. (2015) Clinical application of next-generation sequencing for Mendelian diseases, Human Genomics, 9, article n. 10.).
Dall’utilizzo delle tecnologie NGS per l’analisi di piccoli pannelli di geni in patologie con basi molecolari ben caratterizzate (ad esempio: cromatinopatie, rasopatie, paraparesi spastiche ereditarie), si è passati al sequenziamento NGS di tutti i geni-malattia ad oggi conosciuti (Esoma Clinico), che sono circa 4.000, o addirittura di tutti i geni che compongono il genoma umano (Whole Exome Sequencing, WES, meglio conosciuto come Esoma) (Xue et al. 201510Xue, Y., Ankala, A., Wilcox, W., et al. (2015) Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing, Genetics in Medicine, 17: 444-451.).
La differenza di target tra le due piattaforme citate, Esoma Clinico ed Esoma, conferisce loro potenzialità diverse. Poiché il riscontro di mutazioni patogeniche in geni-malattia noti, analizzati da entrambe le piattaforme, ha valore diagnostico, sia l’Esoma Clinico che l’Esoma possono essere impiegati come test genetici. Solo l’Esoma può, invece, identificare varianti di sequenza con possibile significato clinico in geni non ancora associati a malattie genetiche, consentendo quindi di arricchire le conoscenze nell’ambito della genetica medica, e di delineare nuove associazioni genotipo-fenotipo da inserire nei percorsi clinico-diagnostici.
Considerando che ciascun individuo ha decine di migliaia di varianti nucleotidiche che non hanno conseguenze per la salute, analizzare tutti i geni contenuti nel genoma in un paziente con una patologia genetica, e distinguere la mutazione causativa, comporta uno sforzo interpretativo notevole. Per facilitare il processo identificativo vengono messe in atto diverse strategie, tra cui la contemporanea analisi dei genitori e la consultazione di database pubblici che catalogano le varianti genetiche umane, sia benigne, sia patogeniche (ad esempio: gnomAD, dbSNP e ClinVar; cfr. Sitografia – Alcuni link di riferimento). Un’ulteriore difficoltà riguarda la necessità di dimostrare che la variante, identificata in un gene non associato a patologia, sia effettivamente responsabile del quadro clinico osservato nel paziente in esame. Tramite studi funzionali in vitro e in vivo, è possibile valutare l’effetto della variante sulla struttura/funzione della proteina codificata dal gene in questione, sulla morfologia/omeostasi cellulare, sullo sviluppo/funzionalità di organi e organismi. La condivisione dei dati genetici e clinici anonimizzati con altri laboratori, attraverso piattaforme online dedicate (ad esempio, GeneMatcher), consente in alcuni casi di identificare altri individui non imparentati con fenotipo simile e varianti nello stesso gene, fornendo ulteriori evidenze a supporto di un ruolo del gene stesso nella patogenesi della malattia in esame (Quintáns et al. 201411Quintáns, B., Ordóñez-Ugalde, A., Cacheiro, P., Carracedo, A., Sobrido, M. J. (2014) Medical genomics: The intricate path from genetic variant identification to clinical interpretation, Applied and translational genomics, 3(3): 60-67.).
3.6. Conclusioni
I test genetici costituiscono una componente essenziale del percorso diagnostico di persone affette da patologie genetiche. Sebbene l’identificazione della mutazione patogenica abbia un impatto terapeutico ancora limitato, risulta essere molto importante per la gestione clinica dei pazienti e dei familiari, tanto più se avviene in tempi brevi. L’impiego di nuove tecnologie, oltre a migliorare la sensibilità per l’identificazione di CNVs e varianti di sequenza, ha accorciato notevolmente i tempi necessari per l’esecuzione dei test genetici, accrescendo quindi l’utilità clinica della diagnosi molecolare nell’ambito della genetica medica.
Nel breve futuro nuove tecniche di sequenziamento, capaci di sequenziare frammenti lunghi (long-read, circa 10 kb) e quindi di identificare alterazioni cromosomiche strutturali e CNVs, andranno probabilmente a sostituire la citogenetica classica e l’array-CGH, e affiancheranno l’NGS (sequenziamento short-read, fino a 150 bp) per offrire ai pazienti la possibilità concreta di giungere a una diagnosi definitiva in tempi brevi, sottoponendosi a due soli test in grado di identificare l’intero spettro di alterazioni genetiche e cromosomiche possibili.